Clinical Trial Specimen Transport: Requirements, Regulations, and Best Practices

Every clinical trial depends on the integrity of the biological specimens collected from study participants. When a blood sample, tissue biopsy, or pharmacokinetic draw leaves a clinical site and travels to a central laboratory, the conditions it experiences during transit directly determine whether the resulting data can support a regulatory submission. Clinical trial specimen transport is not a generic logistics task. It is a regulated, protocol-driven process governed by FDA expectations, ICH Good Clinical Practice guidelines, and international dangerous goods regulations. A single transport deviation, whether a temperature excursion, a chain of custody gap, or a documentation failure, can invalidate an entire batch of study data and trigger audit findings that jeopardize the trial itself.
For sponsors, contract research organizations, and clinical sites operating across the Northeast, the logistics complexity multiplies with geography, specimen type diversity, and the volume of concurrent studies. A Phase III oncology trial collecting frozen tumor specimens requires fundamentally different transport infrastructure than a pharmacokinetic study shipping ambient blood draws. What both require is a medical courier built for clinical trials that understands protocol-specific handling requirements, maintains audit-ready documentation, and delivers the regulatory compliance that protects trial integrity from site to laboratory.

1. FDA and ICH GCP Requirements for Specimen Transport
Clinical trial specimen transport operates under a regulatory framework that is substantially more demanding than standard diagnostic specimen logistics. The FDA’s guidance on specimen handling establishes clear expectations for how biological samples must be collected, stored, and transported to ensure data integrity. These expectations apply to all specimen types collected under an Investigational New Drug application, from routine safety labs to specialized biomarker assays. The FDA does not prescribe a single transport method, but it requires that sponsors demonstrate their specimen handling procedures maintain sample quality and produce reliable analytical results.
The ICH E6(R2) Good Clinical Practice guidelines reinforce these requirements by mandating that all clinical trial processes, including specimen logistics, be documented, reproducible, and auditable. Section 4.9 requires investigators to maintain adequate records of the disposition of all trial-related materials, which includes biological specimens collected at the site. When an FDA inspector reviews a clinical site during a routine inspection or a for-cause audit, the transport documentation for every specimen shipment is part of the inspectable record. Gaps in that documentation are classified as observations that can escalate to warning letters if they suggest systemic failures in data integrity.
Sponsors must also comply with NIH clinical research regulations when federally funded trials are involved, adding another layer of oversight to specimen management. The practical implication is that every clinical trial specimen transport event must generate a documented record that proves the specimen was handled according to the study protocol, maintained within specified conditions, and delivered with an unbroken chain of custody.
Regulatory Requirements Include:
- FDA expectation that transport procedures maintain specimen quality sufficient for reliable analytical results
- ICH GCP documentation requirements for the disposition and handling of all trial-related biological materials
- Audit-ready transport records that can withstand FDA inspection at both clinical sites and central laboratories
- Protocol-specific handling instructions that reflect the stability requirements of each specimen type
- Documented corrective actions for any transport deviations, including root cause analysis and preventive measures
2. Chain of Custody for Regulatory Submissions
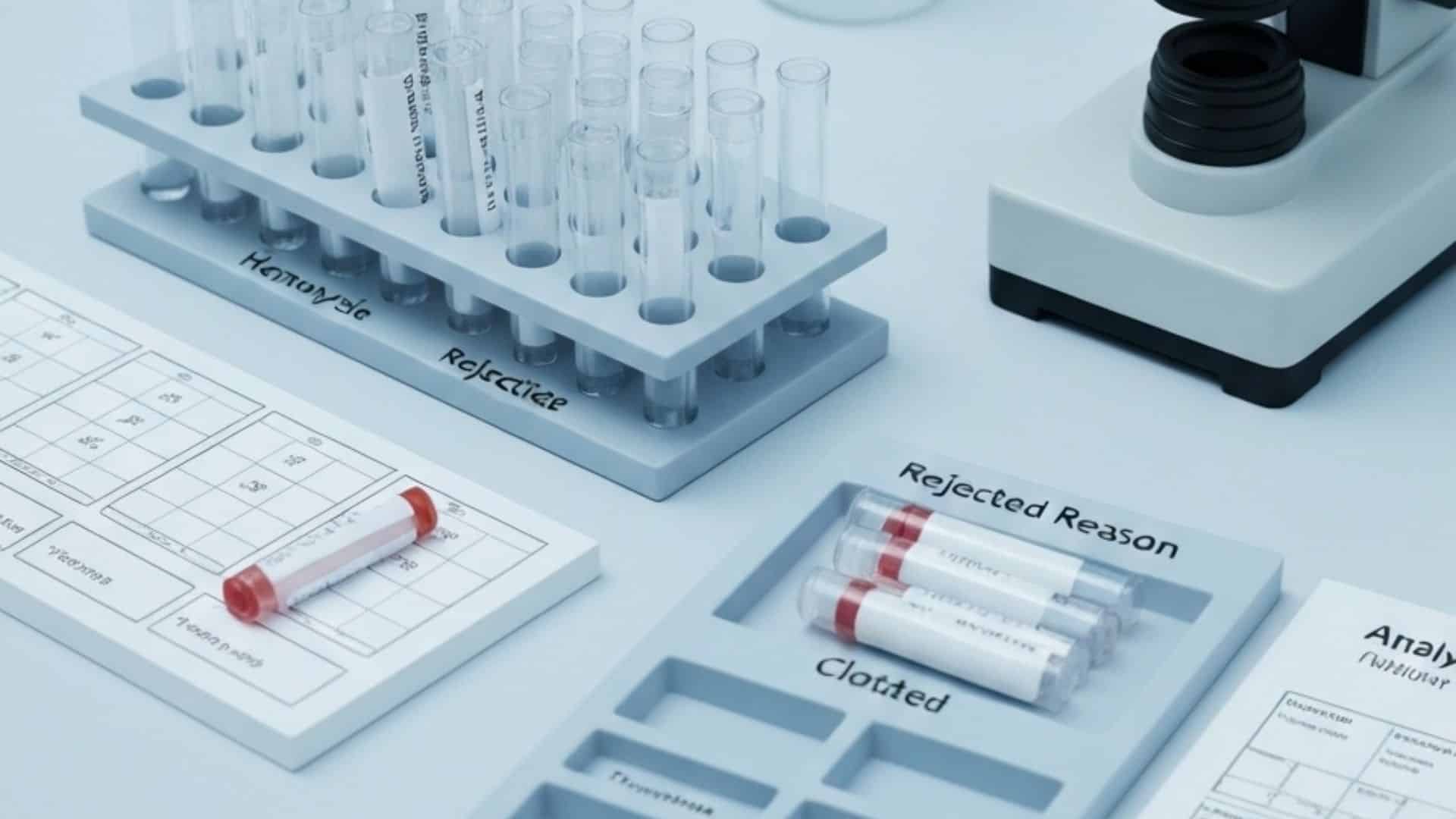
In clinical trials, chain of custody is not just a quality measure. It is a regulatory requirement that directly affects whether analytical data can be included in a New Drug Application or Biologics License Application. The FDA expects sponsors to demonstrate that every specimen used to generate pivotal efficacy or safety data was handled under controlled, documented conditions from collection through analysis. A chain of custody courier service designed for clinical trials must provide documentation that meets this evidentiary standard, not merely confirm that a package was picked up and delivered.
The chain of custody record for a clinical trial specimen must capture the identity of every individual who handled the specimen, the exact time of each transfer, the condition of the specimen at each handoff, and the environmental conditions maintained throughout transit. This level of documentation is what separates clinical-grade chain of custody from standard courier tracking. When an FDA auditor reviews the data package for a pivotal biomarker analysis, they trace the specimen journey backward from the analytical result to the collection event. Any undocumented gap in that journey creates a data integrity question that the sponsor must resolve.
carGO Health’s digital chain of custody platform generates the audit-ready documentation that clinical trial logistics require. With electronic signatures at every handoff, timestamped condition assessments, and real-time delivery tracking that records the complete transport journey, every specimen shipment produces a defensible regulatory record. Across 200,000+ completed orders, this documentation infrastructure has been built to satisfy the most rigorous audit requirements.
Chain of Custody Documentation Must Include:
- Electronic signatures with timestamps at collection, pickup, every intermediate transfer, and final delivery
- Specimen identity verification through barcode scanning that matches the chain of custody log to the protocol visit schedule
- Condition assessment at each handoff documenting specimen integrity, packaging condition, and label verification
- Continuous GPS tracking that documents the transport route and any unplanned stops or deviations
- Environmental monitoring data linked to each specimen’s chain of custody record for integrated traceability
3. Temperature Control Across Multiple Stability Tiers
Clinical trial protocols specify precise temperature requirements for each specimen type, and these requirements vary significantly across studies and even within a single study’s specimen collection schedule. A pharmacokinetic whole blood sample may require ambient transport between 15 and 25 degrees Celsius. Serum samples for immunogenicity testing may require refrigerated transport between 2 and 8 degrees Celsius. Tumor biopsies for genomic analysis may require frozen transport at minus 20 degrees Celsius or cryogenic storage at minus 70 degrees Celsius or below. Each temperature tier demands different packaging, different monitoring systems, and different courier training.
A temperature-controlled medical transport operation for clinical trials must be able to manage all of these tiers simultaneously, often within a single collection visit that generates multiple specimen types destined for different laboratories. The logistics manual for a Phase III trial may specify five or more distinct temperature conditions across its specimen collection schedule. Any temperature excursion that falls outside the protocol-defined acceptable range creates a deviation that must be reported, investigated, and documented, and the affected specimens may need to be excluded from analysis.
carGO Health provides cold chain logistics across all five temperature tiers: ambient, refrigerated, frozen, dry ice, and cryogenic. Continuous temperature monitoring with calibrated data loggers records conditions throughout transit, and automated alerts trigger immediate corrective action when conditions approach excursion thresholds. For biologics and cold chain transport, this monitoring infrastructure is essential to protecting specimen stability and producing the temperature documentation that central laboratories and sponsors require.
Temperature Tier Requirements:
- Ambient (15 to 25 degrees Celsius): routine hematology, clinical chemistry, and pharmacokinetic whole blood draws
- Refrigerated (2 to 8 degrees Celsius): serum immunogenicity samples, certain biomarker assays, and cytokine panels
- Frozen (minus 20 degrees Celsius): plasma aliquots, pharmacodynamic markers, and protocol-specified frozen storage specimens
- Dry ice (minus 70 degrees Celsius): genomic specimens, RNA-based assays, and specimens requiring ultra-low temperature stability
- Cryogenic (minus 150 degrees Celsius and below): viable cell preparations, certain immunology specimens, and long-term biobank storage transfers
- Continuous calibrated monitoring with automated excursion alerts across all temperature tiers
4. Packaging, Labeling, and UN 3373 Compliance
Clinical trial specimens are classified as Category B biological substances under international dangerous goods regulations, which means they must be packaged, labeled, and transported in compliance with UN 3373 requirements. The UN 3373 packaging standard requires a triple-containment system: a leak-proof primary receptacle, a leak-proof secondary container with absorbent material, and a rigid outer shipping container that meets drop-test certification standards. These are not optional quality preferences. They are regulatory mandates enforced by the Department of Transportation under 49 CFR and by IATA dangerous goods regulations when air transport is involved.
Labeling requirements for clinical trial specimens add protocol-specific elements beyond the standard UN 3373 markings. Each specimen must carry the study protocol number, the subject identification number (never the patient name, to protect blinding and privacy), the visit number, the specimen type, the collection date and time, and any special handling instructions. Mislabeling is one of the most common causes of specimen rejection at central laboratories, and it creates documentation gaps that are difficult to resolve retroactively.
For clinical trial specimen transport operations, packaging validation is a critical quality step. Before any specimens are shipped under a new protocol, the packaging configuration must be validated to confirm it maintains the required temperature range for the anticipated transit duration under worst-case conditions. This validation is documented and included in the study’s logistics plan as evidence that the transport system is fit for purpose.
Packaging and Labeling Requirements:
- UN 3373 triple-containment packaging with leak-proof primary and secondary containers and certified rigid outer packaging
- Protocol-specific labeling including study number, subject ID, visit number, specimen type, and collection date and time
- DOT 49 CFR compliance for ground transport and IATA DGR compliance when specimens require air transport
- Packaging validation documented for each protocol to confirm temperature maintenance under worst-case transit conditions
- Absorbent material sufficient to contain the entire volume of all primary receptacles in the event of breakage
5. How Transport Deviations Invalidate Trial Data
The consequences of transport failures in clinical trials extend far beyond a single lost specimen. When a temperature excursion degrades an immunogenicity sample, that data point is excluded from the analysis population. If the excursion affects multiple specimens from the same collection visit across several sites, the missing data can compromise the statistical power of the study’s endpoint analysis. In extreme cases, the FDA may determine that transport-related data quality issues undermine the reliability of the entire dataset, requiring additional studies or delaying approval.
Transport deviations also generate significant operational burden. Each deviation requires a formal investigation, root cause analysis, corrective and preventive action documentation, and notification to the sponsor and potentially the IRB. For CLIA and CAP compliant specimen transport supporting clinical trials, deviation management must be systematic, not reactive. The cost of investigating a single deviation, including staff time, regulatory documentation, and potential protocol amendments, often exceeds thousands of dollars. Multiply that across dozens of sites in a multi-center trial, and transport quality becomes a direct driver of study cost and timeline.
This is why specialized medical courier services for biotech and pharmaceutical companies focus on deviation prevention rather than deviation response. carGO Health’s 98.9% on-time performance rate reflects an operational infrastructure designed to prevent the transport failures that compromise trial data. With 24/7/365 operations, AI-powered dispatch routing, and continuous environmental monitoring, the goal is to eliminate deviations before they occur. Across medical specimen transport operations serving NY, NJ, CT, MA, VT, NH, Eastern PA, DE, MD, and VA, every route is optimized for the specific time and temperature requirements of the specimens it carries.
Consequences of Transport Deviations:
- Specimen exclusion from analysis, reducing statistical power and potentially requiring additional patient enrollment
- Formal deviation investigations requiring root cause analysis, CAPA documentation, and sponsor notification
- FDA audit findings that classify transport failures as data integrity observations, potentially triggering warning letters
- Study timeline delays when critical specimen data is unavailable for interim or final analyses
- Financial impact from deviation investigations, protocol amendments, and potential re-collection requirements
Key Takeaways
Clinical trial specimen transport is a regulated, protocol-driven process that directly affects data integrity, regulatory compliance, and ultimately whether a drug or biologic reaches approval. The requirements are clear: FDA and ICH GCP guidelines demand documented chain of custody, validated temperature control, compliant packaging under UN 3373, and audit-ready records at every stage of the specimen journey. Meeting these requirements is not optional for any organization involved in clinical research.
The cost of getting clinical trial logistics wrong is measured in invalidated data, FDA audit findings, study delays, and the financial burden of deviation management. The value of getting it right is measured in clean datasets, successful regulatory submissions, and trials that stay on timeline and budget. For sponsors, CROs, and clinical sites across the Northeast, carGO Health provides the specialized infrastructure, documentation systems, and operational discipline that clinical trial specimen transport demands. To evaluate how our clinical trial logistics capabilities align with your study requirements, schedule a consultation with our team.
Frequently Asked Questions
What are the FDA requirements for clinical trial specimen transport?
The FDA requires that clinical trial specimen transport procedures maintain sample quality sufficient to produce reliable analytical results. This includes documented chain of custody, validated temperature control, protocol-specific handling procedures, and audit-ready records that demonstrate compliance during site inspections. Sponsors must show that transport conditions did not compromise the integrity of data used in regulatory submissions.
How does chain of custody differ for clinical trial specimens versus diagnostic specimens?
Clinical trial chain of custody must meet a higher evidentiary standard because the resulting data may be submitted to the FDA as part of a drug or biologic approval application. This requires protocol-specific documentation, subject-level traceability linked to the study visit schedule, and records that can withstand regulatory audit. Standard diagnostic chain of custody focuses on CLIA and CAP compliance, while clinical trial chain of custody must also satisfy ICH GCP and FDA inspection requirements.
What temperature ranges are required for clinical trial specimen transport?
Clinical trial protocols specify temperature requirements based on specimen type and analytical method. Common ranges include ambient (15 to 25 degrees Celsius), refrigerated (2 to 8 degrees Celsius), frozen (minus 20 degrees Celsius), dry ice (minus 70 degrees Celsius), and cryogenic (minus 150 degrees Celsius and below). Each protocol defines acceptable ranges, and any excursion outside these ranges constitutes a deviation requiring investigation and documentation.
What is UN 3373 and why does it apply to clinical trial specimens?
UN 3373 is the classification for Category B biological substances, which includes most clinical trial specimens. It requires triple-containment packaging with a leak-proof primary receptacle, a leak-proof secondary container with absorbent material, and a certified rigid outer container. Compliance is mandatory under DOT 49 CFR for ground transport and IATA dangerous goods regulations for air transport. Failure to comply can result in regulatory penalties and specimen rejection.
What happens when a transport deviation occurs during a clinical trial?
A transport deviation triggers a formal investigation process that includes root cause analysis, corrective and preventive action documentation, sponsor notification, and potentially IRB reporting. Affected specimens may be excluded from analysis, reducing the study’s evaluable data. Repeated deviations can result in FDA audit findings classified as data integrity observations, which may lead to warning letters or clinical holds depending on severity and scope.
Related Resources